Targeting in-vivo protein delivery

Recombinant proteins provide a powerful research tool and have also transformed the treatment of many diseases. From the smallest peptide to larger proteins, such as antibodies, how proteins are delivered and reach their target is just as important as their activity on the target.
The physiological effect of recombinant proteins is mediated by binding to proteins present in the target tissue. Recombinant proteins typically need availability to be sustained over weeks and months to achieve an in-vivo effect. However, the poor stability of some proteins provides significant challenges to this outcome. This difficulty is compounded by the ubiquity of many targets throughout the body leading to off-target toxicity. Together, these challenges have hampered the in-vivo application of recombinant proteins
Protein stability
Protein stability is dependent on the structure of a protein and the specific qualities of the environment in which the protein is deployed: High levels of proteases, present in serum or in the extracellular matrix, will reduce stability (other factors present in the serum and the extracellular matrix increase stability). Proteins can also be filtered from fluids e.g. by the kidney removing proteins from sera. Proteins, including some growth factors, can also be taken up and metabolized by cells.
The stability of a naturally occurring protein is an important part of its function. In general, there is an association between the size of a protein and stability. Large proteins such as antibodies tend to be more stable. Also, structural proteins such as collagen, are inherently durable.
Many proteins have short half-lives that create challenges for the in-vivo utility of their recombinant versions.
Addressing protein stability
A wide variety of approaches have been explored to address protein stability. These can be broadly divided into mechanisms that improve the inherent stability of each protein molecule and technologies that provide depot formulations.
Increasing the protein stability of individual molecules
The stability of individual protein molecules can be increased by a variety of mechanisms: For example, joining an unstable bioactive protein with a more stable protein to create a chimeric fusion protein can improve stability. In general, the stable protein partner is a structural protein that doesn’t impact the bioactivity of the unstable protein. Notably, antibody long-chain fragments have been used to create more stable drugs. For example, Enbrel, a blockbuster drug used to treat rheumatoid arthritis, fuses the IgG1 antibody’s heavy chain to stabilize the TNF-α receptor.
Individual molecules may also be conjugated with chemicals such as polyethylene glycol (PEG) to increase stability. There are around 30 approved protein drugs that utilize PEGylation to improve stability.
Specific modifications of amino acid sequences can also increase stability. For example, LongR3-IGF is a stabilized form of IGF-1 which is stabilized by specific amino acid substitutions as well as an extended carboxy terminus.
Whilst stabilizing proteins is a useful strategy for increasing the in-vivo utility of proteins, the increased durability that can be provided may not be sufficient to achieve the desired outcome. Moreover, increasing the stability of individual molecules may lead to increased path lengths (the distance an individual molecule is able to travel before it degrades) which promotes diffusion to adjacent off-target tissues.
Depot formulations
An alternative to improving the stability of each individual protein molecule per se is the use of depot formulations. These may be nanoparticles but are typically micron-scale structures (>500 nm) or larger. Multiple copies of the protein are held within each depot in a stabilizing matrix which also provides a sustained release mechanism. With depots, the protein is highly stable whilst held within the depot and then degrades once released. Lantus is a very successful insulin depot formulation where the insulin has amino acid modifications that alter its pH-dependent solubility. The drug is formulated to be soluble at the pH of the storage buffer. However, the drug crystallizes when injected into blood. The crystals then slowly dissolve sustainably releasing bioactive insulin.
Depot formulation
Nanoparticles
On a scale intermediate between free molecules and microparticles, the term “nanoparticles” or "nanocarriers" covers a myriad of synthetic and natural materials (
Toxicity
When used in-vivo, the off-target effects of proteins can generate toxicity. For example, antibodies targeted against checkpoint inhibitors, such as PD-L1, now widely used to treat cancer, generate serious side effects in about 40% of patients treated. This is because as well as the target protein in the tumor microenvironment (TME), the drug binds to copies of the protein outside the TME disturbing the fine balance of the immune system.
Injection site and stability
Proteins are degraded in the digestive tract necessitating parenteral delivery for in-vivo applications. The precise mode of delivery modulates subsequent biodistribution and stability. As well as the assault of proteases, the metabolism of proteins by cells and their filtration by organs such as kidneys affects protein durability and distribution. For example, interleukin 2 (IL-2), used to treat some types of cancer by upregulating the activity of T cells, has a half-life of a few minutes if injected intravenously due to the filtering effect of the kidney on circulating blood. However, if IL-2 is injected intra-peritoneally, the bolus of the drug is protected from the filtering effects of the kidney and is slowly released into the bloodstream. This effectively increases the half-life to several hours.
Localized delivery
Like all drugs, proteins require release kinetics that keeps their concentrations high enough for efficacy and low enough to be tolerated. The ratio of these two parameters provides a therapeutic index. The larger the ratio, the better. To achieve a useful therapeutic index many in-vivo applications of recombinant proteins necessitate localized delivery. For example, protein therapeutics that enable bone regeneration must be targeted to the specific area of bone. Even when delivered locally a protein provided at high doses a protein can seep to non-target areas. Spinal fusion cages used to fuse vertebrae to treat back pain can be functionalized with BMP-2, a growth factor that promotes bone growth. However, using conventional BMP-2 at very high doses to offset BMP-2’s short half-life, resulted in many patients suffering debilitating and painful ossification of surrounding musculature.
Combining a protein into a structure such as a hydrogel creates a depot formulation that slowly releases the protein improving release kinetics. Polylactic-co-glycolic acid (PLGA), the most widely studied and clinically validated hydrogel for this purpose, achieves sustained release but also suffers from “burst release” of cargo proteins. Burst release is a by-product of the PLGA microsphere manufacture: when a protein is incorporated into PLGA (which can be a technically challenging process due to the denaturation of the protein) surface tension effects cause the protein to accumulate at the surface of the PLGA structures. Consequently, there is an initial burst of protein availability, typically 25-30% of the total loaded, which leads to dangerously high concentrations.
In-vivo uses of recombinant proteins
Because proteins are large molecules, they are not ordinarily able to gain access to intracellular targets. Therefore, the in-vivo application of proteins is largely restricted to extracellular targets. Therapeutically, these are either cell membrane proteins such as receptor proteins or secreted proteins, such as antibodies, cytokines, chemokines and other protein messenger molecules.
Targeted delivery
Protein delivery systems have been developed to impact the biodistribution of proteins following delivery. The simplest solution is localized administration i.e. direct injection into the target tissue. However, for disseminated diseases, such as cancer, multiple sclerosis and infectious diseases, localized administration may not possible.
To increase the utility of proteins, efforts are being made to target drugs (both protein and small molecule) to sites of diseases. This is most commonly achieved using antibodies that bind to the surface of specific target cells. This is known as active targeting. However, as has been pointed out by others this is a misnomer since the antibody is not actively guided to a target. Rather, it is an affinity-based binding. Typically, the protein is intravenously injected, disperses throughout the body in the circulatory system and accumulates at the target by binding to a surface epitope recognized by the antibody.
Trojan horse delivery
A mechanism for actually delivering a protein to target tissue and improving over-dispersed delivery may be provided by immune cells which actively seek out disease. Protein-containing particles taken up by phagocytic immune cells can be carried a site of disease where they are recruited into the tissue under chemotactic cues provided by the chemokines and cytokines secreted by inflamed tissue. This strategy has the advantage that the drug is actively carried into diseased tissue beyond the limits of capillaries rather than just diffusing.
The possibility of using phagocytic immune cells to transport particles to the site of disease is sometimes referred to as a Trojan horse drug delivery. Although this is an idea that was proposed over 40 years ago, the concept has been difficult to reduce to practice. Trojan horse delivery is often referred to as passive targeting (compared with active targeting provided by antibodies). However, considering that the phagocytic cells are actively recruited into the inflamed tissue under the guidance of chemotactic proteins, perhaps it is better described as active targeting?
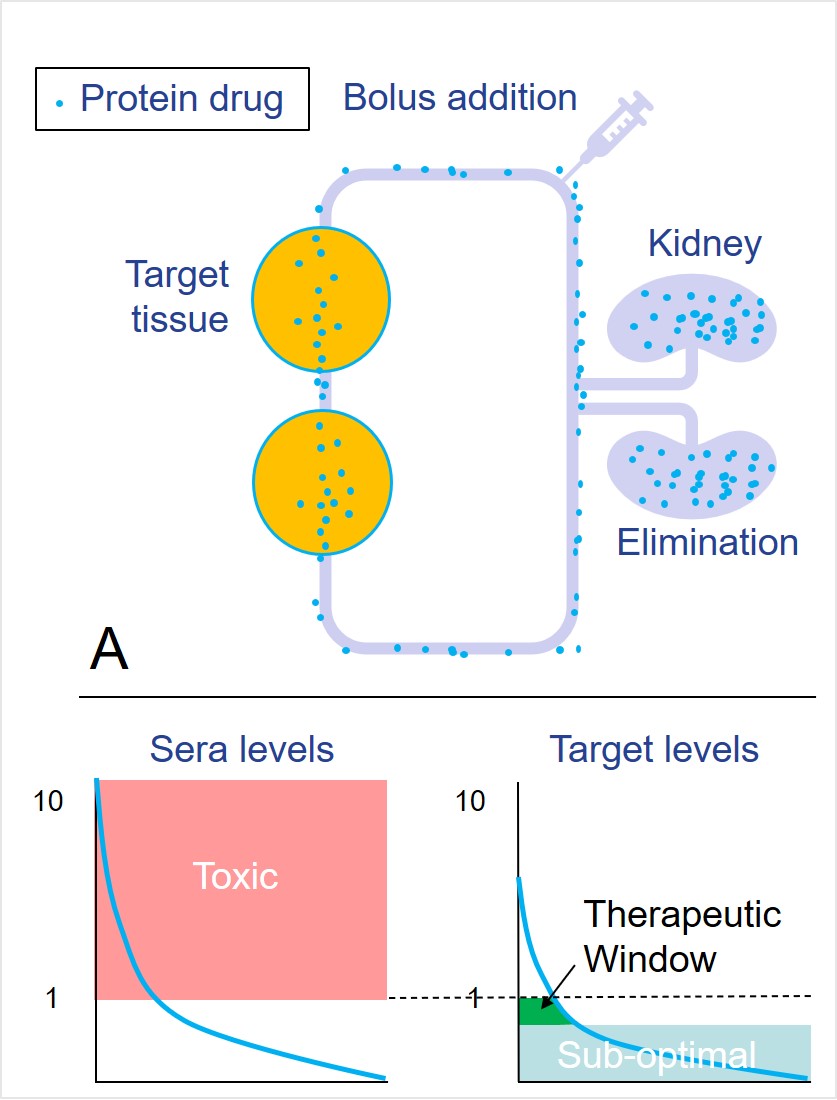
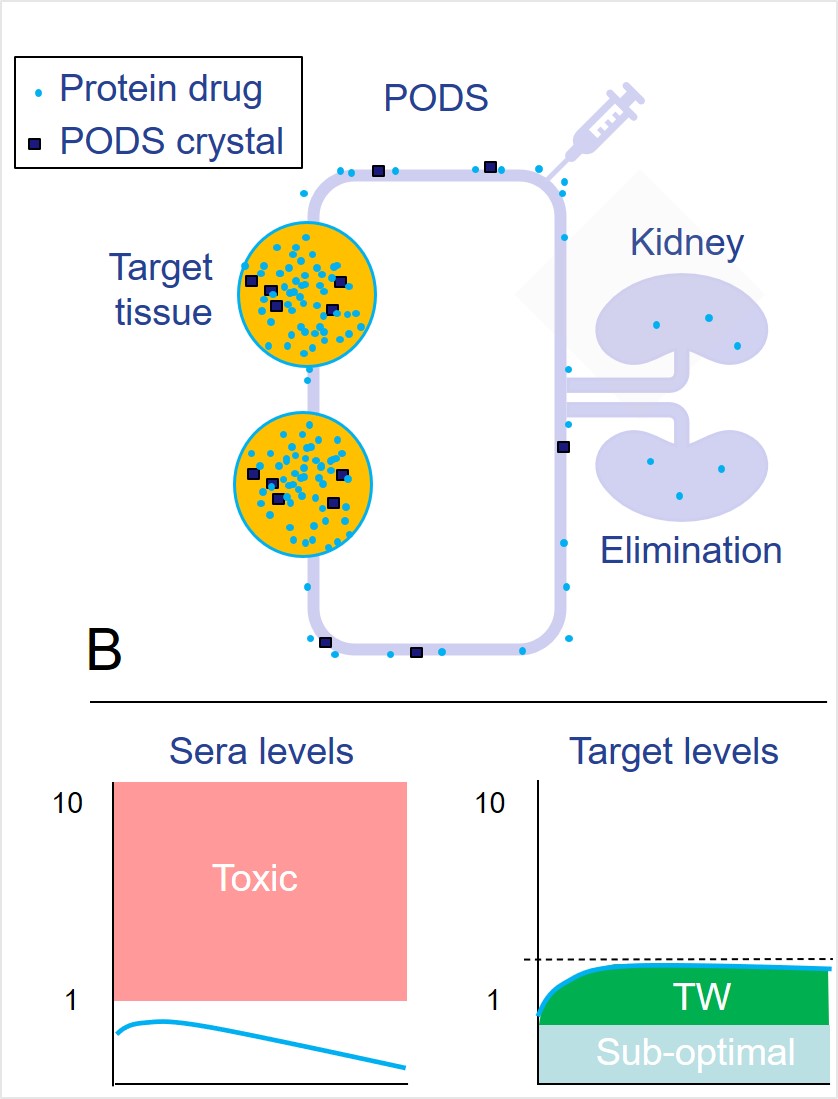
Biodistribution predicted following intravenous injection of (A) conventional protein/nanoparticles and (B) proteins released from a phagocytosed depot (PODS crystals). Use of PODS crystals results in a significantly increased therapeutic window.
Self-assembling protein carriers
The POlyhedrin Delivery System (PODS®) has been used in a variety of in-vivo applications to provide targeted delivery and sustained release of proteins. PODS is a technology is built around the unique properties of the polyhedrin protein. When expressed at high levels, the polyhedrin protein self-assembles into characteristic micron-scale cubic PODS crystals (PCs). These cubes range in size from 200 nm up to 7 µm spanning the realm of nanoparticles and microparticles.
Co-expression of a secondary cargo protein fused with an affinity tag which binds to the polyhedrin protein results in the incorporation of the cargo protein to form a co-crystal. These crystals are robust, durable and non-brittle and can withstand considerable physical stress. Since these crystals are composed entirely of protein, they are degraded by proteases. This results in the steady release of cargo proteins over a period of 1-2 months.
Similar to silk proteins which are widely used in medical applications, polyhedrin crystals appear to be weakly immunogenic and are well-tolerated.
The physical characteristics of PCs make them highly efficient for phagocytic ingestion although this can be ameliorated by secondary encapsulation into hydrogel microspheres. Phagocytic ingestion of PCs with subsequent release of bioactive cargo molecules to modulate adjacent heterogenous cells has been demonstrated.
In-vivo applications of PODS
Use of PODS crystals as a cell-implant survival agent
A common challenge with cell therapy is the poor survival and maturation of implanted cells. This attrition reflects differences between the in-vitro and in-vivo environments that the implanted cells have to adapt to. As many cells are growth factor-dependent in culture, recombinant growth factors implanted alongside the implanted cells have been shown to enhance cell survival. Eventually, the cells become adapted to their new environment and the growth factors are no longer needed. To be effective, these growth factors must be implanted alongside the cells and retain bioactivity for an extended period (days to weeks) whilst the implanted cells to adapt. Conventional growth factors with their short half-lives retain bioactivity for too-short a period to be useful.
PCs have been shown to be effective in providing sustained release of cell survival agents and have been used to enhance the survival of neuronal cells implanted in the back of the eye and inner ear in mouse models.
Survival of auditory neurons is enhanced by PCs
During sensorineural hearing loss, the destruction of hair cells in the cochlea occurs. Since these cells are a major source of neurotrophic factors, this causes the death of afferent auditory neurons. Brain-derived neurotrophic factor (BDNF) has been shown to promote survival and neuronal differentiation of spiral ganglion neurons both in vitro. However, in vivo effects of conventional BDNF are limited by a short half-life. The harsh, nutrient-deprived environment of the cochlea is a major challenge for the survival of implanted cells. To promote the in vivo survival and neuronal differentiation of transplanted human Embryonic Stem Cell (hESC)-derived Otic Neuronal Progenitors (ONPs), Akira Matsuoka and colleagues utilized BDNF-containing PCs as a long-term neurotrophic growth factor delivery system. These promoted neurite extension towards the bony wall of the cochlea during a 90-day post-transplantation period.
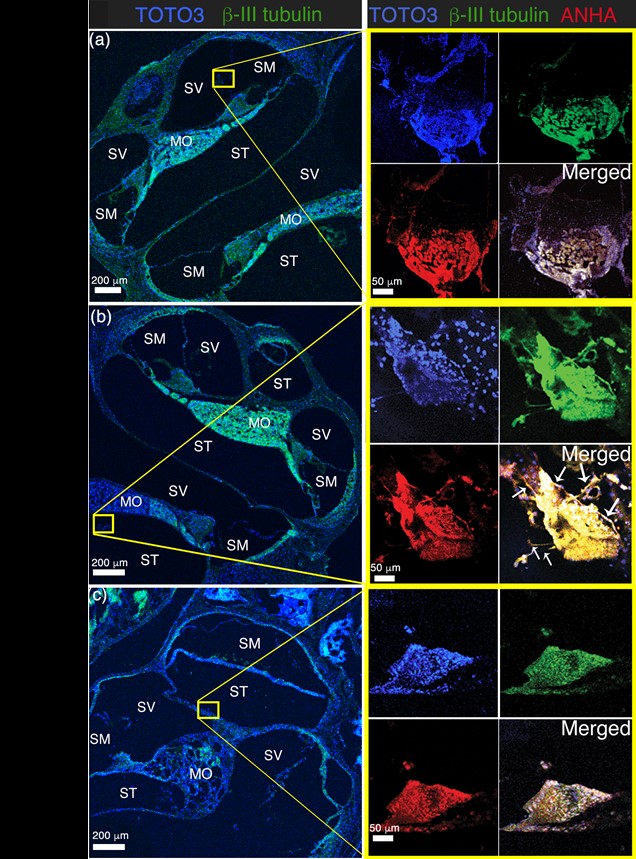
PODS® hBDNF treatment supports the long-term in vivo survival and neuronal differentiation of hESC-derived ONP cells transplanted in DTR+ mice cochlea. Immunohistochemistry of transplanted hESC-derived ONP spheroids in the DTR+ mouse cochlea with (a) GrowDex®-T (only) or (b) PC hBDNF/GrowDex®-T. Left panels: Low magnification (10X) microphotographs of the DTR+ mouse cochlea. Each yellow square indicates the anatomical location of a transplanted hESC-derived ONP spheroid. Right panels: High magnification (40X) microphotographs of transplanted hESC-derived ONP spheroids stained with nuclear marker TOTO3 (blue), β-III tubulin (green), and AHNA (red). Small white arrow: neurites. SM: scala media, SV: scala vestibuli, ST: scala tympani, MT: the middle turn of the cochlea, and MO: modiolus. (Data courtesy of Akihiro Matsuoka, Northwestern University)
Retinal ganglions
PCs containing BDNF, as well as glial-derived neurotrophic factor (GDNF), were also used to enhance transplant survival of dissociated retinal ganglion cells (RGCs). Dissociated RGCs were transplanted by intravitreal injection into mouse eyes either in the presence or absence of PC- BDNF and PC- GDNF. Two weeks following implantation, eyes were dissected and stained with Thy1 and β3-tubulin to reveal implanted cells (green) against the background of existing neuronal cells (red). Not only was the number of cells which survived significantly higher with the PCs present, the maturation of the cells was remarkably improved with axon extensions more pronounced. Further information on this experiment is available in an Application Note.
![]()
Dissected eyes, two weeks post-RGC transplantation. Thy1 was used to detect RGCs and β3-tubulin (TUJ-1) used as general neuronal marker. (A) without PCs (B) with PCs Significantly improved maturation with increased levels of axon growth (arrows) was achieved in the PC-treated eye. [Data courtesy of Petr Baranov, Harvard University)
Localized therapeutic delivery
PCs have been successfully used in a variety of experimental in-vivo settings where localized sustained release is required to regenerate damaged tissue. As well as increasing simplicity and improving efficacy, the fewer interventions required with PCs has animal welfare benefits.
Bone regeneration
The use of bone morphogenic proteins (BMPs), notably BMP-2 and BMP-7, to accelerate bone remodelling is well established. BMPs have widespread effects on cells outside the bone. Consequently, their delivery needs to be localized. The use of very high levels of BMP has been used to address the short half-life. But such a brute-force approach can lead to complications (see above). PCs provide an ideal solution to this problem. This has been demonstrated using a rat calvaria model of bone remodelling in which collagen pads infused with either conventional BMP-2 or PC-BMP-2. Vehicle only or empty PCs (no cargo molecule) were included as controls. In this study, PCs significantly outperformed conventional BMP-2 leading to the remodelling of bone to native levels.
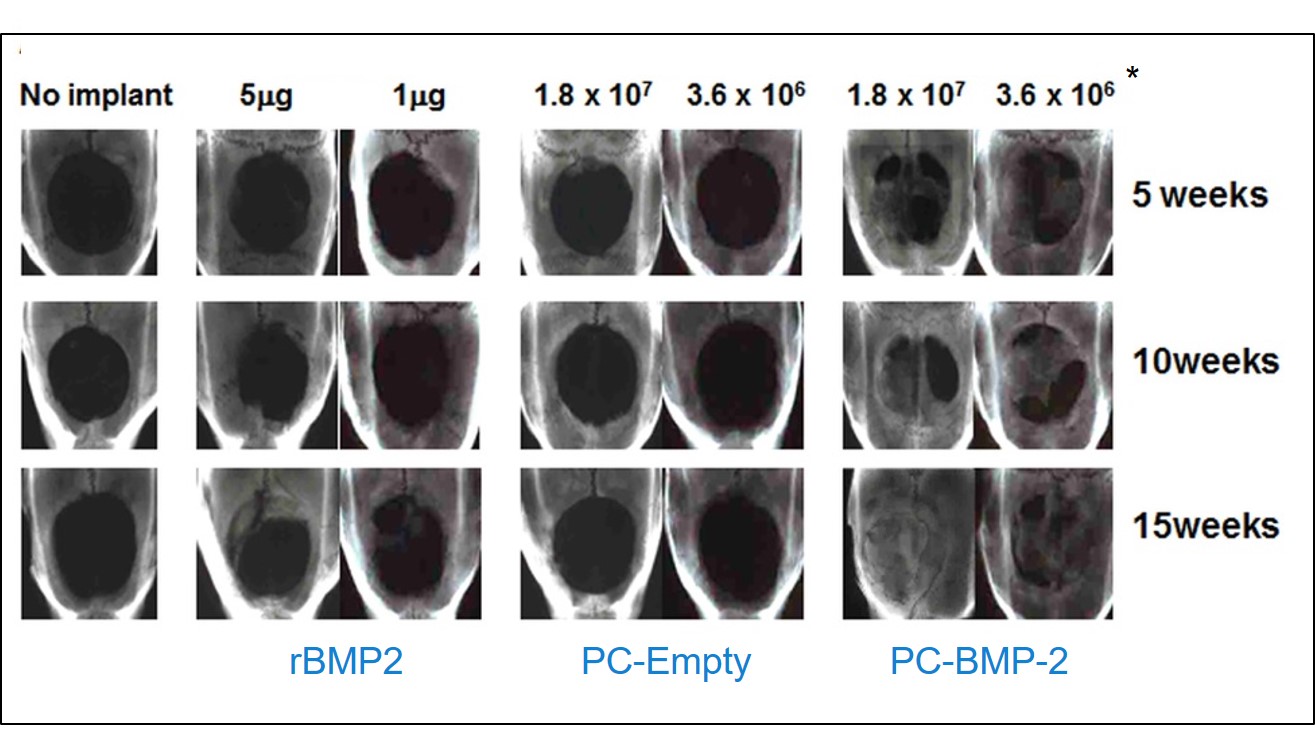
Regeneration of bone in a rat calvaria model. Collagen sponges functionalized with conventional recombinant BMP-2 or empty PCs or PCs were inserted into defects generated in rat calvaria bone. Only PC-BMP-2 generated good bone remodelling. (Data courtesy Hajime Mori, Kyoto Institute of Technology)
Cartilage regeneration
Damage to the cartilage within joints is associated with the development of osteoarthritis (OA). By repairing this cartilage damage, it may be possible to slow or even reverse the course of OA. In a mouse model of OA, both PC-BMP-2 and PC-BMP-7 accelerated the healing of a defect in murine knee articular cartilage.
Targeted systemic delivery
For disseminated diseases including metastatic cancer, only systemic drug delivery can be expected to reach all sites of disease. Localized delivery (e.g. intra-tumoral injection) is not a viable option. However, this can lead to widespread off-target toxicity impacting healthy tissues. The key to drug delivery for cancer treatment is to accumulate sufficient levels of the drug within the tumor microenvironment whilst suppressing drug levels in healthy tissue.
Nanoparticles and microparticles have the potential to modulate the delivery of proteins due to the way they interact with target cells and immune cells. Such particles can be made from a wide range of synthetic and natural materials. The size of a particle is a major factor impacting distribution. The size of PCs (0.4 – 6 µm) makes them highly amenable to phagocytic uptake. This opens up the possibility of using a Trojan horse (see above) whereby phagocytic cells (primarily monocytes, and macrophages) actively transport PCs (and their protein cargo) to areas of inflammation and disease. To be effective, this approach would require large numbers of cells to transport the drug. Fortunately, in cancer as much as 50% of the cells in the tumor microenvironment (TME) are macrophages. These have a lifespan of about 2 weeks and are constantly being replaced with newly arriving macrophages. Another potential advantage of this approach derives from monocytes' ability to cross the blood-brain barrier.
Significant progress has already been made in translating this approach into the clinic. Macrophages that have ingested PCs remain viable and it has been demonstrated that PCs are able to secrete their cargo in a bioactive form to impact nearby cells.
Whilst PCs will accumulate in the TME, any PODS that remain in sera only slowly release their cargo. As sera is constantly cleared by the kidney, the cargo fails to accumulate limiting serum-toxicity. Thus, the interaction of PCs with phagocytic cells produces a very striking shift in the biodistribution of proteins.
In soon-to-be-published work using mouse models of cancer, it has been shown that intravenously injected PCs are effective against cancer whilst maintaining very low, almost undetectable, serum levels.